Photochemistry and Excited States
Excited electronic states and nonadiabatic processes are key to many phenomena. We explore quantum modeling combined with molecular dynamics to address complexity and improve calculations for photochemistry, also in condensed phase.
Excited electronic states and nonadiabatic processes are vital for photochemistry, energy conversion, and applications like photodynamic (cancer) therapy. Modeling these phenomena requires simultaneous treatment of electronic and nuclear degrees of freedom. Due to computational complexity, we usually rely on a semiclassical approach: treating the electronic part quantum mechanically to obtain energies and forces, which evolve the nuclei through ab-initio molecular dynamics. Our focus is on nonadiabatic dynamics in the condensed phase, going beyond standard approaches (like continuum models or QM/MM) by treating the solvent environment explicitly at the same level of theory.
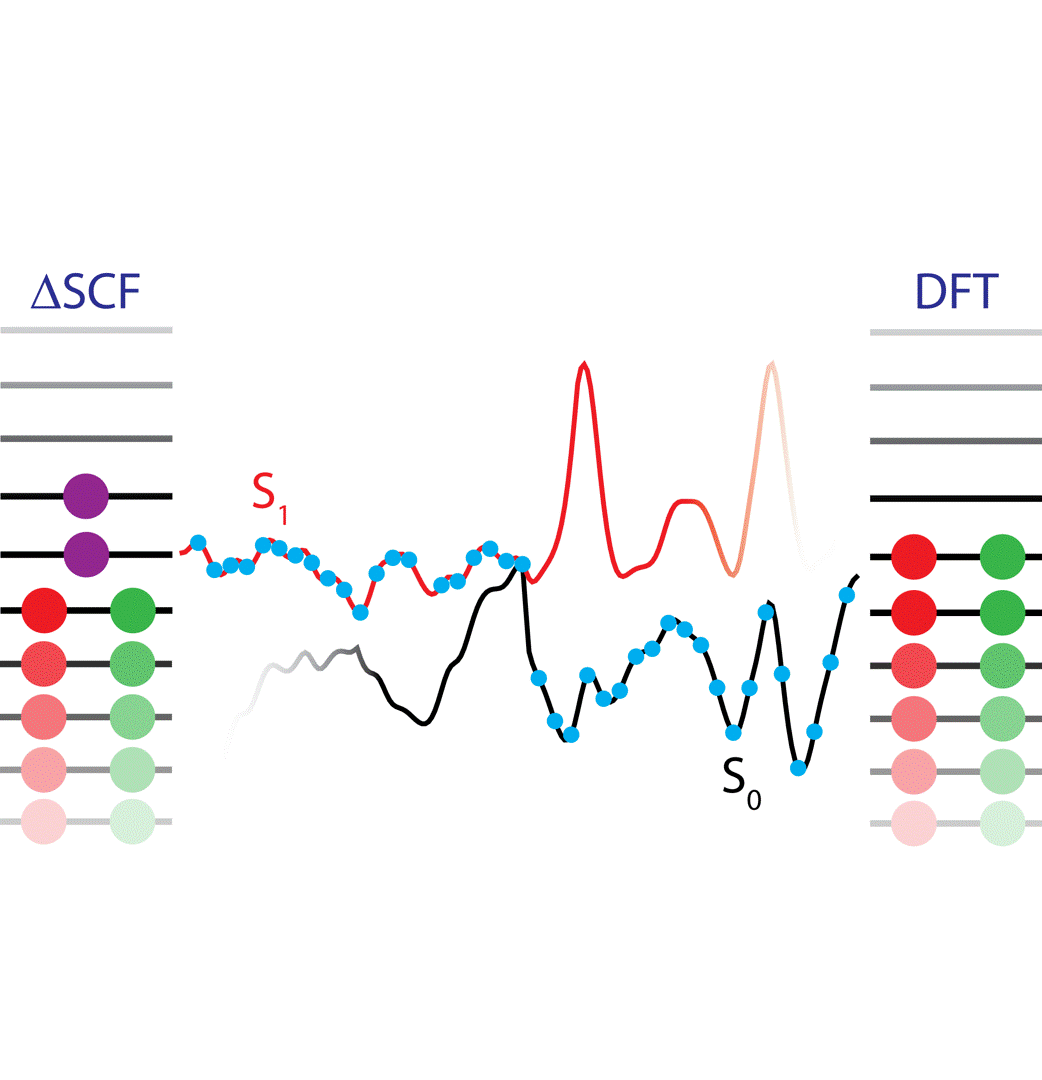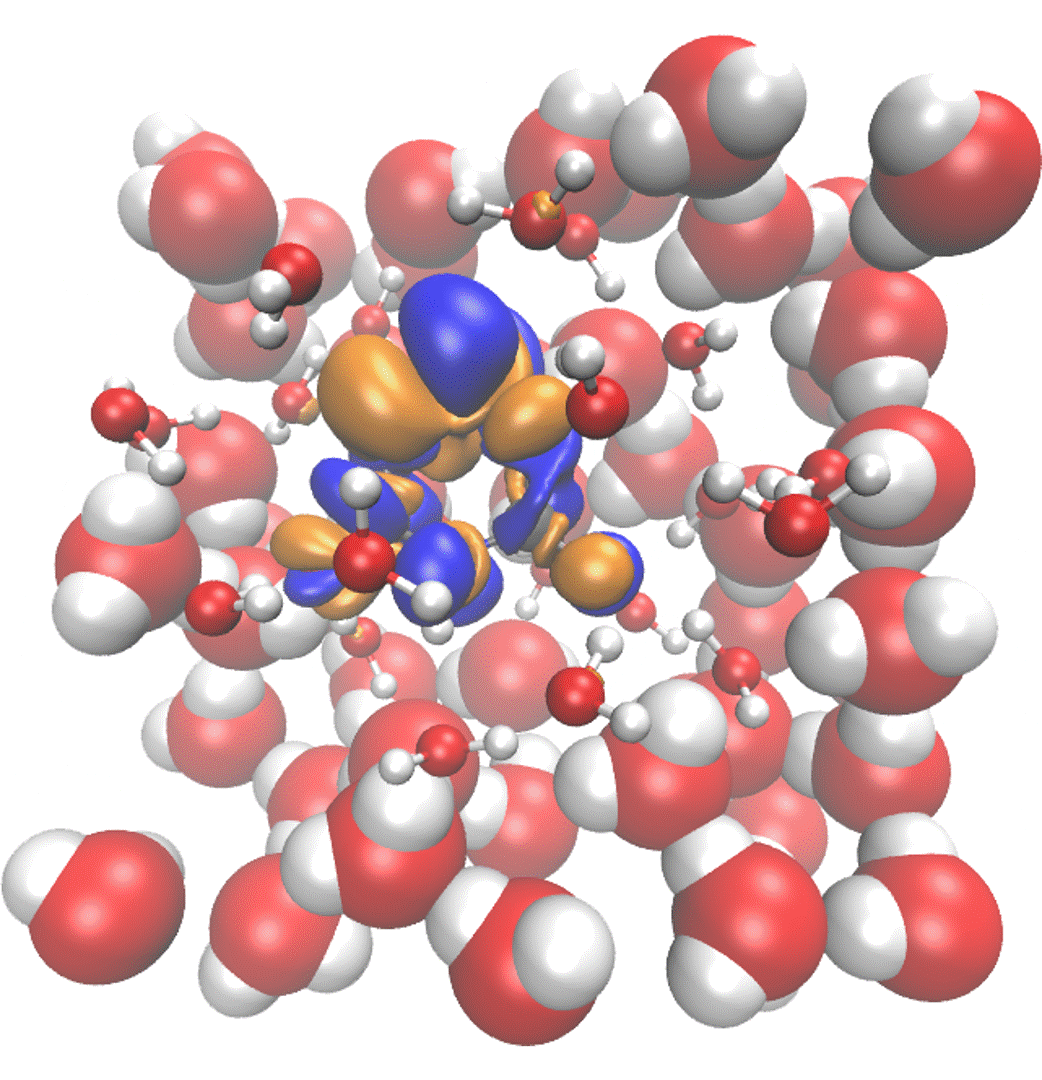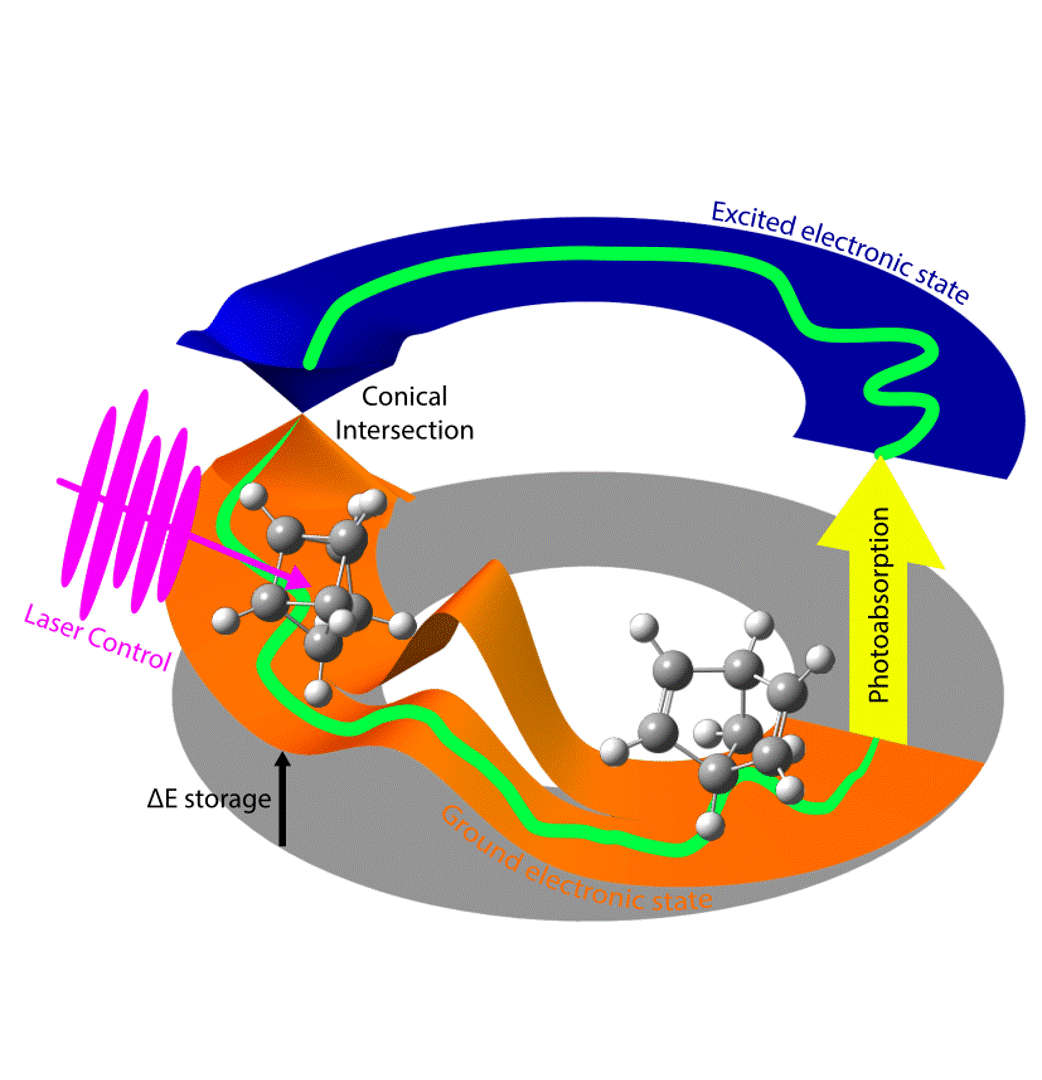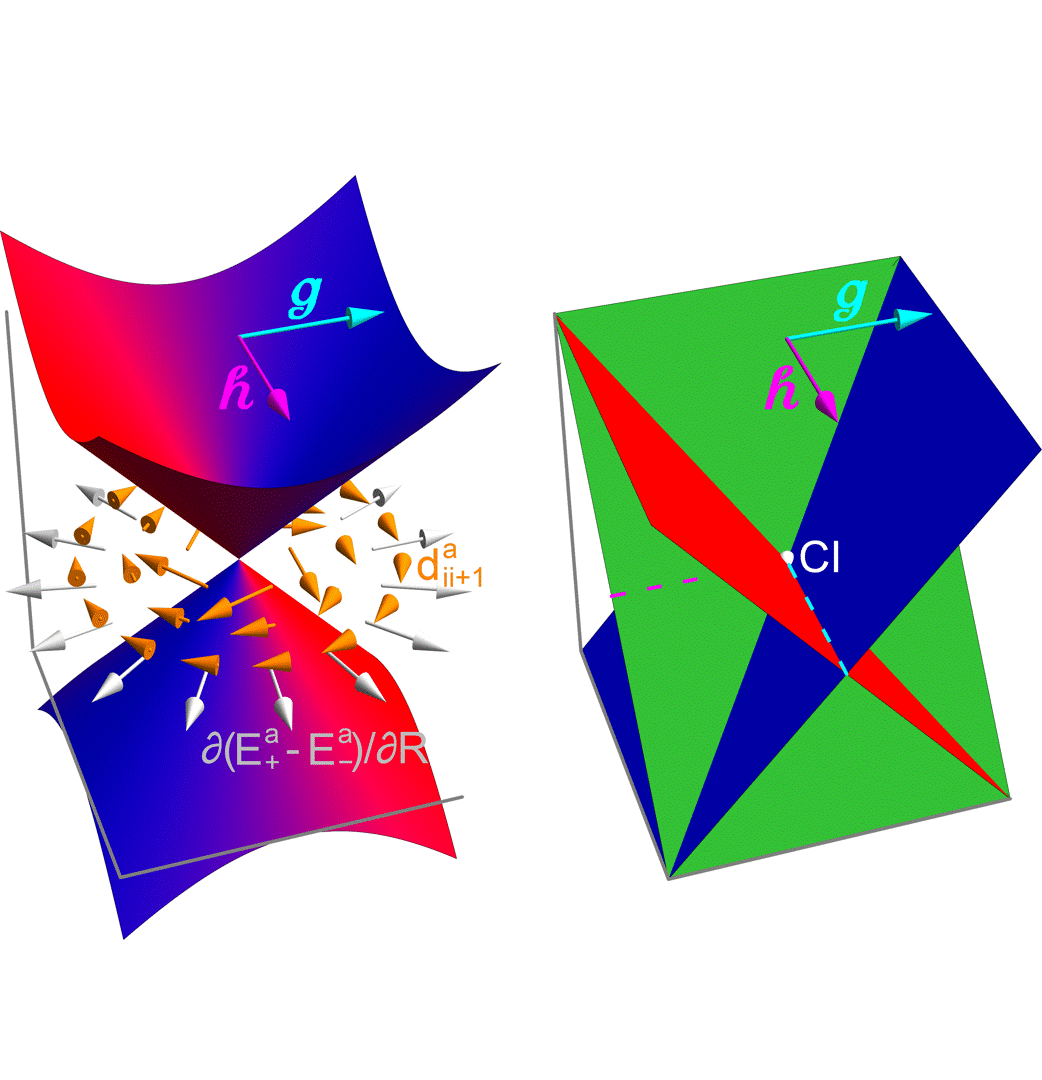
The ΔSCF method is an attractive tool for studying excited electronic states by optimizing the electron density for one or more specific excited states. We have worked, among others, on improving ΔSCF convergence and related property calculations as well as accessing higher excited states and going beyond the single-reference picture. Another direction has concerned the advancement of efficient (spin flip) time-dependent density functional theory (TDDFT) and real time TDDFT approaches for large gas and condensed phase systems.
Selected Publications
L. Hernandez-Segura, S. Luber
Spin-Flip TDDFT within the Sternheimer Formulation: A Gaussian and Plane Wave Implementation
J. Phys. Chem. A, 2025, 129, 42, 9798-9809
K. Hanasaki, T. de Jong, K. Komarov, R. Kumar, M. Mališ, J. Mattiat, L. Hernandez-Segura, L. Schreder, A. Sinyavskiy, S. Luber
Exploring Excited-State Electronic Structure, Spectroscopy, and Nonadiabatic Dynamics with CP2K’s Multifaceted Approach
J. Phys. Chem. A, 2025, 129, 32, 7313-7344
K. Hanasaki, S. Luber
Development of Real-Time TDDFT Program with k-Point Sampling and DFT + U in a Gaussian and Plane Waves Framework
J. Chem. Theory Comput., 2025, 21, 4, 1879–1891
E. Vandaele, M. Mališ, S. Luber
A Local Diabatisation Method for Two-State Adiabatic Conical Intersections
J. Chem. Theory Comput. 2024, 20, 2, 856–872
L. Schneider, M. Kalt, S. Koch, S. Sithamparanathan, V. Villiger, J. Mattiat, F. Kradolfer, E. Slyshkina, S. Luber, M. Bonmarin, C. Maake, B. Spingler
BODIPY-Based Photothermal Agents with Excellent Phototoxic Indices for Cancer Treatment
J. Am. Chem. Soc. 2023, 145, 8, 4534–4544
M. Mališ, E. Vandaele, S. Luber
Spin-Orbit Couplings for Nonadiabatic Molecular Dynamics at the ΔSCF Level
J. Chem. Theory Comput. 2022, 18, 7, 4082-4094
C. Kumar, S. Luber
Robust ΔSCF calculations with direct energy functional minimization methods and STEP for molecules and materials
J. Chem. Phys. 2022, 156, 154104
E. Vandaele, M. Mališ, S. Luber
The ΔSCF method for non-adiabatic dynamics of systems in the liquid phase
J. Chem. Phys. 2022, 156, 130901
E. Vandaele, M. Mališ, S. Luber
The photodissociation of solvated cyclopropanone and its hydrate explored via non-adiabatic molecular dynamics using ΔSCF
Phys. Chem. Chem. Phys. 2022, 24, 5669-5679
M. Mališ, S. Luber
ΔSCF with Subsystem Density Embedding for Efficient Nonadiabatic Molecular Dynamics in Condensed-Phase Systems
J. Chem. Theory Comput., 2021, 17, 3, 1653-1661
M. Mališ, S. Luber
Trajectory Surface Hopping Nonadiabatic Molecular Dynamics with Kohn– Sham ΔSCF for Condensed-Phase Systems
J. Chem. Theory Comput., 2020, 16, 7, 4071-4086